Ideally, there should be an early diagnosis, this performed via a non-invasive technique and before the beginning of the genital virilization of the fetus. Diaz A, Laufer MR Breech LLAA of PC on A, Care AC of O and GC on AH. J Clin Endocrinol Metab. Consultation with an endocrinologist is recommended for patients who require complex hormone regimens. (2006) 91:420514. By contrast to childhood, in adolescence, longer-acting steroids are often used and regimens of 5 mg of prednisolone or 0.25 mg of dexamethasone are recommended (53). Most patients may seek medical advice at any stage of their life due to symptoms related to androgen excess. This content is owned by the AAFP. Pediatr Endocrinol Rev. The classical form of the disease, occurs in 1 out of 16,000 live births worldwide. Horm Res. The condition is not identified on routine infant blood screening and usually becomes evident in late childhood or early adulthood. (1991) 56:63540. Aldosterone also may be low, which leads to dehydration and low sodium and high potassium levels. doi: 10.1530/EJE-11-0019, 76. Females with NCCAH who desire to conceive should be aware of the risk of giving birth to an infant with the classical form of the disease. Another important consideration regards the techniques used for 17 OHP evaluation. Both classic and nonclassic forms of the disease are caused by deficiencies in the adrenal enzymes that are used to synthesize glucocorticoids. (2003) 349:77688. Merke DP. Front. Its DNA sequence can be altered by at least nine mutations, many of which leave the enzyme impaired but not totally inactive.2,3,6, The incidence of classic congenital adrenal hyperplasia is especially high in Madagascar and certain areas of Alaska. It should be noted that 17 OHP values are crucial for the diagnosis, but not helpful during followup. Speiser PW, et al. (2013) 78:197203. Classical congenital adrenal hyperplasia is rare, affecting only one in 14,000 patients, but mild forms of the disease may occur in one of every 100 to 1,000 persons.1,2 The condition is caused by a deficient synthesis of cortisol; most cases are related to 21-hydroxylase or 11- hydroxylase deficiency35 (Figure 1). (2004) 33:97104. Mayo Clinic. Treatment should be initiated immediately on confirmation of pregnancy.4,10 Dexamethasone readily crosses the placenta and suppresses the fetal adrenal gland. Rev Endocr Metab Disord. The adrenal glands produce important hormones, including: In people who have CAH, a gene change (mutation) results in a lack of one of the enzymes needed to make these hormones. Pubic hair and other signs of puberty may appear at a very early age. If you have concerns about your child's growth or development, make an appointment with your child's health care provider. Excess androgen. J Clin Endocrinol Metab. In patients with inadequate cortisol secretion after stimulation, or if OCPs and antiandrogens cannot be tolerated, a course with GCs is highly recommended (57). (2005) 63:2704. doi: 10.1067/mob.2000.108020, 13. In ovarian ultrasound, 77% of them displayed polycystic ovaries and 41% increased ovarian size.

(2007) 90:41421. During major life-threatening stress, surgery, or serious illness, patients with NCCAH who are glucocorticoid-treated may require larger or more frequent doses of glucocorticoids given that their adrenal function is iatrogenically suppressed. Acne is reported in almost 33% of NCCAH subjects (36). According to the Endocrine Society guidelines, NCCAH patients should be given the option to discontinue GC therapy when symptoms resolve (60). Kazmi D, Bailey J, Yau M, Abu-Amer W, Kumar A, Low M, et al. Finally, based on data collected by Speiser et al., 9% of individuals with NCCAH displayed 17 OHP values lower than 2 ng/ml (that corresponds to 6nmol/L). Perched on top of each of your kidneys, your adrenal glands produce hormones that help regulate your metabolism, immune system, blood pressure and other essential functions. Women with mild congenital adrenal hyperplasia often present with hirsutism, oligomenorrhea or infertility. Moran C, Azziz R, Carmina E, Dewailly D, Fruzzetti F, Ibaez L, et al. Further, in the case of discontinuation, patients should be informed about the possibility of infertility and should be encouraged to seek medical advice if they wish to conceive (19). Pediatrics. https://www.uptodate.com/contents/search. The treatment decision should be based on assessment of the facts and should follow an individualized approach. Int J Pediatr Endocrinol. The earliest case of NCCAH reported is a 6 month old girl who developed pubic hair (11). doi: 10.1210/jcem.86.1.7131, 68. Bonfig W, Bechtold S, Schmidt H, Knorr D, Schwarz HP.
Likewise, the incidence in France and Switzerland ranges between 1:15,472 and 1:23,000 and 1:10,749 and 1:11,661 when the different methods of screening are used (4). doi: 10.2169/internalmedicine.49.3573, 77. doi: 10.1210/jcem-63-2-418.
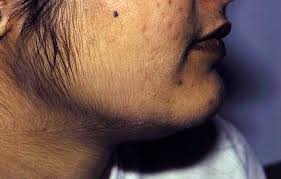
Certainly, the study by Witcel et al. Hence, the use of ACTH stimulated 21-deoxycortisol either singularly or as part of a steroid ratio or steroid profile, may facilitate the biochemical identification of heterozygotes in the future, particularly as LC-MS/MS becomes more widely available [8385]. Chrousos GP, Loriaux DL, Mann DL, Cutler GB. This site complies with the HONcode standard for trustworthy health information: verify here. Ambroziak U, Kepczynska-Nyk A, Kuryowicz A, Wysouch-Cieszynska A, Maunowicz EM, Bartoszewicz Z, et al. All Rights Reserved. Late-onset 21-hydroxylase deficiency mimicking idiopathic hirsutism or polycystic ovarian disease. J Clin Endocrinol Metab. Maintenance therapy is generally achieved with hydrocortisone, in a dosage of 6 to 25 mg per m2 per day given in two to three divided doses.1,3,4,8 Hydrocortisone is preferred over other glucocorticoids because it is short acting and can be given in pulses that mimic natural cortisol secretion. (2013) 27:144851. However, adolescent patients frequently do not show sufficient compliance with chronic administration of drugs and often omit doses. Saygili F, Oge A, Yilmaz C. Hyperinsulinemia and insulin insensitivity in women with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency: the relationship between serum leptin levels and chronic hyperinsulinemia. Merke DP, et al. doi: 10.1210/jc.2009-2631, 20. Additionally, the administration of 2.57.5 mg of prednisolone, a dose considered as normal, exerts a longstanding negative impact on bone metabolism (45). Lastly, given that many therapeutic issues related to the appropriate management of these patients have not as yet been elucidated, it is very important for the attending physician to keep up to date with all developments in this field and to integrate the new data into his clinical practice. Those who survive to adulthood experience premature puberty. The nonclassic form of CAH doesn't cause adrenal crisis. Endocrinol. If you're thinking of starting a family and you're at risk of having a child with CAH, your health care provider may recommend that you see a genetic counselor. They also had significantly increased bisexual or homosexual orientation. J Clin Endocrinol Metab. They were also characterized by significantly higher levels of testosterone, free testosterone, and 17 OHP than patients with other hyperandrogenic syndromes. During this period, the glucocorticoids used are prednisone or hydrocortisone. Deoxycortisol and its metabolites have mineralocorticoid properties and may cause hypertension when they accumulate.2,3,7 Thus, simple blood pressure measurements may help determine the underlying type of congenital adrenal hyperplasia. Clin Endocrinol. (2010) 2010:625105. doi: 10.1186/1687-9856-2010-625105, 9. Very low levels of cortisol in the blood can cause diarrhea, vomiting, dehydration, low blood sugar levels, seizures and shock. One hundred years of congenital adrenal hyperplasia in Sweden: a retrospective, population-based cohort study. This form of glucocorticoid is favored due to its metabolization by placental enzyme 11 OH steroid dehydrogenase II, which prevents glucocorticoid from having any effect on the fetus (71). (2005) 90:331825. An update on prenatal diagnosis and treatment of congenital adrenal hyperplasia. Whether this exposure of the fetus to dexamethasone for preventive measures is medically and ethically acceptable remains controversial (70). Glucocorticoid precursors accumulate in these persons and are converted to androgenic steroids, causing shortened stature, early puberty, severe acne, and virilization and infertility in females.2,3,5,6 Mineralocorticoid synthesis can also be affected, resulting in electrolyte disturbances, hypotension and syncope.5,6, Ninety percent of patients with congenital adrenal hyperplasia have 21-hydroxylase deficiency.24,6 Because this enzyme functions in both glucocorticoid and mineralocorticoid synthesis, some patients with 21-hydroxylase deficiency have insufficient amounts of cortisone and aldosterone (Figure 2). However, it should be mentioned that the perceived incidence of the disease is related to the screening method used. (2008) 37:8599. 1. Signs and symptoms of CAH vary, depending on which gene is affected and the level of enzyme deficiency. The prevalence of alopecia also appeared to increase with age, from 6% in patients during the second decade of their life to 19% in the fifth, indicating again the progressive nature of the disease (39). Among 161 women with NCCAH, presenting symptoms were hirsutism (78%), menstrual dysfunction (54.7%), and decreased fertility (12%) (34). Turcu AF, Auchus RJ. Received: 09 January 2019; Accepted: 22 May 2019; Published: 06 June 2019. Kruse B, Riepe FG, Krone N, Bosinski HAG, Kloehn S, Partsch CJ, et al. In the young female, 2 years post menarche and if normal ovulatory cycles have been recorded, a patient-centered approach toward the hyperandrogenic symptoms that may appear is highly recommended. Mutations of CYP21A2 with minimal (C) or moderate (NC) residual enzyme activity (14, 68, 69). Clinical manifestations and diagnosis of classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency in infants and children. In a multicenter study, the most common symptoms among adolescent and adult women were hirsutism (59%), oligomenorrhea (54%), and acne (33%). J Clin Endocrinol Metab. Speiser PW, Azziz R, Baskin LS, Ghizzoni L, Hensle TW, Merke DP, et al. (2006) 91:26. Furthermore, definitive recommendations regarding situations when genetic testing is not required are difficult given the imperfect genotype-phenotype correlation, particularly for milder mutations [81]. NCCAH is considered as the more frequent and milder form of CAH because of retention of 2050% enzyme activity.
acth concentrations logarithmic Factors that increase the risk of having CAH include: People who have classic CAH are at risk of adrenal crisis. New MI. New MI, Abraham M, Yuen T, Lekarev O. Known gene abnormalities include insertions, deletions, missense/nonsense codons, and point mutations. 2020; doi:10.1056/NEJMra1909786. Takasu N, Nakachi K, Higa H. Development of Graves' hyperthyroidism caused an adrenal crisis in a patient with previously unrecognized non-classical 21-hydroxylase deficiency. 21-Hydroxylase-deficient nonclassic adrenal hyperplasia is a progressive disorder: a multicenter study. Gn EN, Ozn ZA, Alikaifoglu A, Engiz O, Bulum B, Kandemir N. (2011). doi: 10.3109/09513590.2014.994599, 23. For affected adolescent and adult women, it is crucial to investigate any suspicion of NCCAH and determine a firm and accurate diagnosis. Mayo Clinic. Ethnic-specific distribution of mutations in 716 patients with congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. However, the boundaries across the different forms of CAH are not strictly defined, which tends to increase the challenges associated with this disorder. These include premature pubarche, rapid growth, hirsutism, acne, menstrual irregularities, or infertility, as well as many other less prominent manifestations. (2006) 64:211. Birnbaum MD, Rose LI. Hum Mutat. NCCAH patients who are diagnosed during childhood with signs of PP may be treated with hydrocortisone with the aim of suppressing the adrenal hormones and preventing rapid advancement of bone age that could affect final height. Mol Genet Metab. (2009) 5:4908. Lteif AN (expert opinion). There is no known way to prevent CAH. doi: 10.1111/cen.12935, 22. Br J Dermatol. Verma S, Green-Golan L, VanRyzin C, Drinkard B, Mehta SP, Weise M, et al. Gidlf S, Falhammar H, Thiln A, von Dbeln U, Ritzn M, Wedell A, et al. Arch Sex Behav. J Eur Acad Dermatol Venereol. Prevalence of non-classical congenital adrenal hyperplasia due to 21-hydroxylase deficiency in Greek women with acne: a hospital-based cross-sectional study. Dewailly D, Vantyghem-Haudiquet MC, Sainsard C, Buvat J, Cappoen JP, Ardaens K, et al. In our experience, the administration of bicalutamide has achieved significant improvement in cases of severe acne, but similar results were not obtained in cases with severe hirsutism. Once the diagnosis of NCCAH has been established, several issues related to subsequent treatment recommendations warrant consideration. Of note, in children referred with PP, according to different studies, the incidence of NCCAH ranges from 5 to 30% (20, 27). doi: 10.1210/jc.75.6.1421, 44. (2001) 86:20713. Cuhaci N, Aydin C, Yesilyurt A, Pinarli FA, Ersoy R, Cakir B. Nonclassical congenital adrenal hyperplasia and pregnancy. Simonetti L, Bruque CD, Fernndez CS, Benavides-Mori B, Delea M, Kolomenski JE, et al. As a result, genetic testing of the partners of these women is essential to assess the risk of giving birth to a child with the classical form of CAH (64, 67). According to the current guidelines, the above-described therapy is cautiously recommended only for those cases in which the predicted height SD is 2.25 < the target height or even lower (53). doi: 10.1210/jc.2007-2053, 17. No use, distribution or reproduction is permitted which does not comply with these terms. For those women with symptomatic hyperandrogenism or with reported infertility but who wish to conceive, GC therapy is highly recommended. However, we should be very cautious in the interpretation of this test, since it has not been validated in other studies. A single copy of these materials may be reprinted for noncommercial personal use only. (2016) 2016:17. doi: 10.1186/s13633-016-0035-5, 47. (2015) 82:5439. The primary goal of treatment should be the patient's relief from the hyperandrogenic symptoms. Of note, the screening and diagnostic thresholds for LC-MS/MS quantified 17 OHP are yet to be defined. All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication. Am J Obstet Gynecol. Most crucially, meanwhile, the exposure of a young organism to a very potent GC during a particularly sensitive period of fetal programming and growth, which might well prove useless in the case of a fetus with the XY karyotype, is not at present supported by robust and unquestionable data. Trakakis E, Papadavid E, Dalamaga M, Koumaki D, Stavrianeas N, Rigopoulos D, et al. 1998-2022 Mayo Foundation for Medical Education and Research (MFMER). Kohn B, Levine LS, Pollack MS, Pang S, Lorenzen F, Levy D, et al. However, due to the perplexity of the disease and its multifaceted nature, there are no specific guidelines for the timing of regimen changes or cessation of glucocorticoid therapy in children. Nebesio TD, Renbarger JL, Nabhan ZM, Ross SE, Slaven JE, Li L, et al. This enzyme is required by the body to make proper amounts of hormones. Eur J Endocrinol. Hydrocortisone is typically used in children, as it most closely resembles the natural hormone (cortisol), but it is not considered a suitable approach in adolescents and young females due to the need for multiple daily dosing. (2012) 25:101721. Prenatal androgenization affects gender-related behavior but not gender identity in 5-12-year-old girls with congenital adrenal hyperplasia. However, most women with NCCAH seek medical assistance when they experience symptoms of androgen excess and, when clinical suspicion prompts testing, elevated basal 17 OHP levels will more likely than not point to a diagnosis of NCCAH. On the other hand, in cases where severe hirsutism and/or acne are present, prescription of oral contraceptives and/or antiandrogens may be advisable. Table 1. Of course, further studies are needed to verify or reject this finding. Accessed Dec. 8, 2021. McCann-Crosby B, Chen M-J, Lyons SK, Lin Y, Axelrad M, Dietrich JE, et al. In some cases, due to advanced bone age and/or severe signs of hyperandrogenism, initiation of hydrocortisone treatment prepubertally may be considered. Gopal-Kothandapani JS, Petkar A, O'Shea E, Banerjee I. Perianal hair as an unusual presentation of non-classical congenital adrenal hyperplasia. NEW The Essential Diabetes Book - Mayo Clinic Press, Mayo Clinic on Incontinence - Mayo Clinic Press, NEW Mayo Clinic on Hearing and Balance - Mayo Clinic Press, FREE Mayo Clinic Diet Assessment - Mayo Clinic Press, Mayo Clinic Health Letter - FREE book - Mayo Clinic Press, Mayo Clinic Graduate School of Biomedical Sciences, Mayo Clinic School of Continuous Professional Development, Mayo Clinic School of Graduate Medical Education, Book: Mayo Clinic Family Health Book, 5th Edition, Newsletter: Mayo Clinic Health Letter Digital Edition. Premature closure of the epiphyses results in short stature even though these children grow at an accelerated rate when young. Meyer-Bahlburg HFL, Dolezal C, Baker SW, New MI. In such an instance regular follow-up is needed. In the presence of hyperandrogenic symptoms, a patient-oriented approach is highly recommended, focusing on the main complaints of the patient. Hence all patients with NCCAH should be offered genetic counseling and molecular assessment of reproductive partners. BMJ Case Rep. (2013) 2013:bcr2013010123. If the fetus is male, dexamethasone therapy can be stopped until after the infant is born. Congenital adrenal hyperplasia (CAH) encompasses a family of autosomal recessive disorders characterized by mild to acutely impaired cortisol synthesis due to a deficiency in one of the five adrenal steroidogenic enzymes required for cortisol production (1, 2). doi: 10.1210/jc.2018-01865, 61. Congenital adrenal hyperplasia in adults: a review of medical, surgical and psychological issues. Any use of this site constitutes your agreement to the Terms and Conditions and Privacy Policy linked below. These data, in conjunction with a few studies that indicate the presence of insulin insensitivity, suggest an unfavorable metabolic profile that could be pathophysiologically explained by the effect of elevated androgen concentrations and/or as a result of glucocorticoid therapy (42, 43). (2010) 49:1395400. doi: 10.1111/j.1365-2265.2004.02106.x, 56. Cortisol may be the only hormone that's deficient.

indicate that irregular menses and acne are reversed within 3 months after the initiation of the glucocorticoid therapy, whereas hirsutism requires nearly 30 months (59). (1985) 37:65067. The urinary opening (urethra) and the vagina may be only one opening instead of two separate openings. Int J Endocrinol Metab. (1994) 78:2416. 17 OHP levels are measured by a variety of immunoassay methods, but as has recently been shown, the most accurate and reliable results were achieved by the implementation of the combination of liquid chromatography with mass spectrometry (LC-MS/MS). Consensus statement on 21-hydroxylase deficiency from the European Society for Paediatric Endocrinology and the lawson wilkins pediatric endocrine society. Even normotensive patients with 21-hydroxylase deficiency (Figure 2) may have improved adrenal suppression with the addition of the aldosterone analog fludrocortisone (Florinef) at dosages of 0.05 to 0.2 mg per day to their regimen. (2015) 145:22636. Miller WL. doi: 10.1210/jc.2006-1757, 63. doi: 10.1210/jcem-55-5-817, 12. However, in our data derived from 280 subjects with the disease, six patients (2.1%) had a baseline 17 OHP value < 6 nmol/L (14). Ogilvie CM, Crouch NS, Rumsby G, Creighton SM, Liao L-M, Conway GS. Mineralocorticoid therapy is not required in any of the cases, as these patients have normal aldosterone secretion and do not develop salt-wasting (58). Bidet M, Bellann-Chantelot C, Galand-Portier M-B, Tardy V, Billaud L, Laborde K, et al. They must also know that the probability that their child will suffer from the classical form of the disease is high, despite treatment. doi: 10.1002/humu.23351. J Clin Endocrinol Metab. Therefore, hydrocortisone should be considered the best form of treatment in cases of GC supplementation therapy. Merke DP, Poppas DP. Most importantly, it should be clarified to parents that dexamethasone administration does not modify patient status but is directed toward reducing the need for surgery rather than preserving life or intellectual capacity. Lancet Diabetes Endocrinol. Carmina E, Lobo RA. Genetics and clinical presentation of classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Premature adrenarche: etiology, clinical findings, and consequences. The clinical expression of NCCAH is characterized by a high level of polymorphism as concerns not only age of onset but also the different signs and symptoms. Azziz R, Hincapie LA, Knochenhauer ES, Dewailly D, Fox L, Boots LR. The preferred GC treatment in children is usually hydrocortisone 1015 mg/m2, divided into three doses. J Clin Endocrinol Metab. Gastaud F, Bouvattier C, Duranteau L, Brauner R, Thibaud E, Kutten F, et al. Signs and symptoms of classic CAH may include: Atypical genitalia. Escobar-Morreale HF, Sanchn R, San Milln JL. doi: 10.1007/s12020-015-0656-0, 33. Ovarian suppression reduces clinical and endocrine expression of late-onset congenital adrenal hyperplasia due to 21-hydroxylase deficiency. have in several studies reported impaired psychological profile among patients with NCCAH due to 21OH deficiency. Diagnosis of congenital adrenal hyerplasia in fetuses that are at risk for congenital adrenal hyperplasia can be determined using human leukocyte antigen haplotype or by demonstration of excess cortisol precursors in amniotic fluid. (2010) 73:43644. doi: 10.1210/jcem.78.2.8106606, 8. can lead full lives.
{kind=link}